




















What is the MDR (Medical Device Regulation) in Europe, and how does it affect the industry?
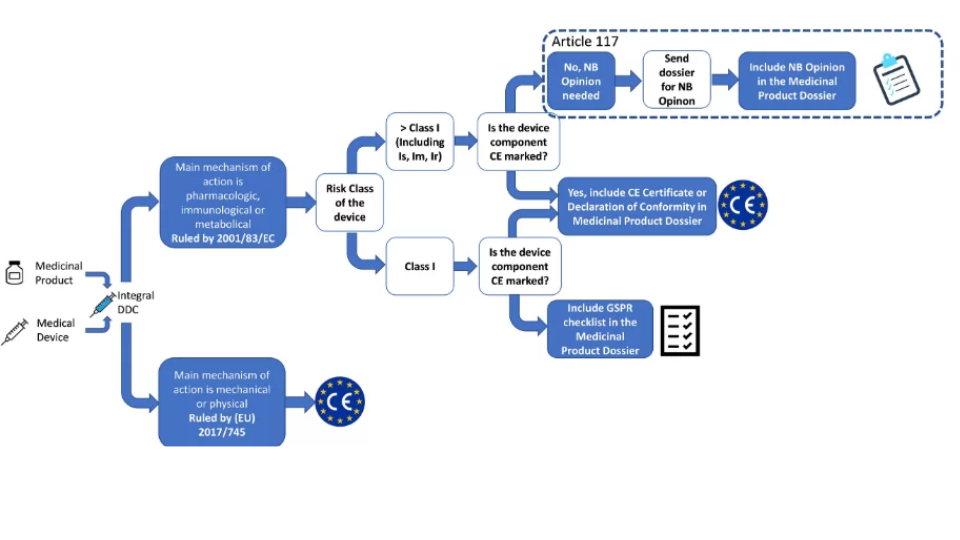
The Medical Device Regulation (MDR) is a comprehensive regulatory framework enacted by the European Union to replace the Medical Device Directive (MDD) and the Active Implantable Medical Devices Directive (AIMDD). The MDR, which officially came into effect on May 26, 2021, is designed to ensure the safety, quality, and performance of medical devices marketed within the EU. It significantly impacts the medical device industry by imposing stricter requirements and enhancing oversight.
Key Features of the MDR:
- Expanded Scope:
- Covers a broader range of devices, including software, certain aesthetic products, and devices with no intended medical purpose (e.g., colored contact lenses).
- Includes new classification rules for software as a medical device (SaMD).
- Stricter Classification Rules:
- Devices are subject to more rigorous classification criteria, leading to higher-risk classifications for many products.
- This affects the regulatory pathway and documentation requirements.
- Enhanced Safety and Performance Requirements:
- Focus on clinical evaluation and post-market surveillance (PMS).
- Devices must demonstrate safety and efficacy through clinical data that meet stricter standards.
- Unique Device Identification (UDI):
- Mandates the implementation of a Unique Device Identification system to improve traceability throughout the supply chain.
- Notified Bodies and Certification:
- Notified Bodies must be re-designated under MDR, reducing the number of authorized entities available for certification.
- All devices must be recertified under MDR, regardless of prior approvals under MDD.
- Eudamed Database:
- Establishes a centralized European database for medical devices to enhance transparency.
- Includes data on clinical investigations, PMS, certifications, and vigilance reporting.
- Post-Market Surveillance and Vigilance:
- Manufacturers must implement robust PMS systems and conduct periodic safety updates.
- Vigilance requirements ensure prompt reporting of adverse events and corrective actions.
- Stricter Labelling Requirements:
- Devices must include detailed information for end-users, ensuring clarity and safety.
Impact on the Medical Device Industry:
- Higher Costs:
- Increased regulatory demands lead to higher compliance costs, particularly for SMEs (Small and Medium Enterprises).
- Costs associated with clinical trials, documentation, and UDI implementation have risen.
- Strain on Notified Bodies:
- A limited number of designated Notified Bodies under MDR has led to bottlenecks, delaying certifications and market entry for some devices.
- Disruption for Legacy Devices:
- Devices previously certified under MDD must be reassessed, leading to potential discontinuation of older products that cannot meet MDR standards.
- Encouragement of Innovation:
- While demanding, the MDR pushes manufacturers to develop safer and more effective products with robust clinical evidence.
- Innovators focusing on compliance early in the design process gain a competitive advantage.
- Market Access Challenges:
- Non-EU manufacturers face additional hurdles in aligning with MDR requirements, such as appointing an authorized EU representative.
- Increased Transparency:
- Eudamed and UDI requirements provide greater transparency and accountability, benefiting patients and healthcare providers.
- Focus on Post-Market Activities:
- Enhanced PMS requirements ensure continuous monitoring of devices after approval, reducing risks and improving device reliability.
- Consolidation in the Industry:
- Smaller companies struggling with the cost and complexity of MDR compliance may merge with larger entities or exit the market.
- Longer Time to Market:
- The need for more comprehensive clinical data and stricter Notified Body scrutiny extends the timeline for bringing new products to market.
Benefits of the MDR:
- Patient Safety: Improves the safety and performance of devices available in the EU.
- Market Confidence: Strengthens trust in medical devices through transparency and traceability.
- Alignment with Modern Practices: Addresses advancements in technology, such as AI and digital health, and emerging trends like cybersecurity.
Challenges and Industry Adaptation:
- The industry has had to invest heavily in compliance resources, training, and systems.
- Companies are prioritizing key products for MDR certification, potentially reducing their portfolios.
- Collaboration with regulatory consultants and Notified Bodies has become essential for navigating the transition.
The MDR represents a paradigm shift in the European medical device regulatory landscape. While it introduces challenges, such as increased costs and longer approval times, its focus on safety, transparency, and innovation ultimately strengthens the industry. Companies that embrace these changes and integrate compliance into their strategic planning are better positioned for success in the evolving regulatory environment.








{kind=link}