

















What is the approval process to bring a medical device to market in the India?
The approval process to bring a medical device to market in India is regulated by the Central Drugs Standard Control Organization (CDSCO) under the Medical Device Rules, 2017. It involves a series of steps, from classification to licensing, based on the risk category of the device. Below is a detailed outline of the process:
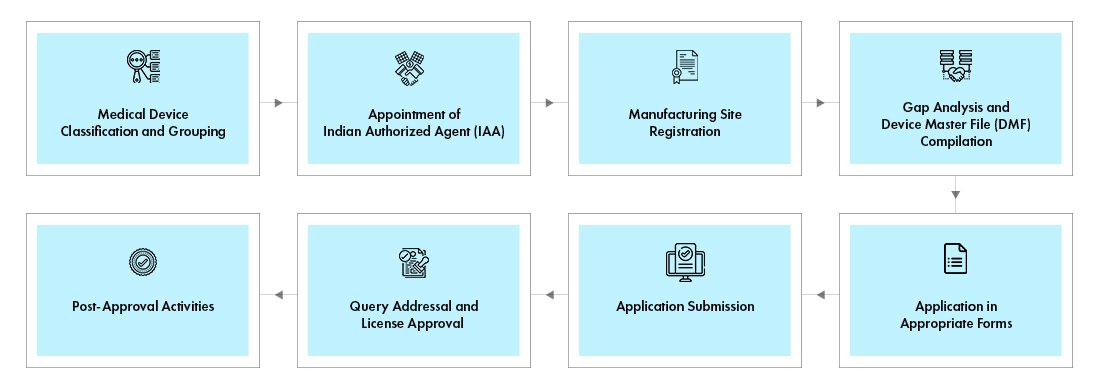
1. Classification of the Medical Device:
The first step is to determine the classification of the device based on its risk level:
- Class A: Low risk (e.g., tongue depressors, surgical dressings).
- Class B: Moderate risk (e.g., infusion sets, diagnostic X-rays).
- Class C: High risk (e.g., orthopedic implants, blood glucose monitors).
- Class D: Very high risk (e.g., pacemakers, heart valves).
Risk classification determines the regulatory pathway and level of scrutiny.
2. Appointment of an Authorized Indian Representative (AIR) (for foreign manufacturers):
If the manufacturer is not based in India, an Authorized Indian Representative must be appointed to act on behalf of the manufacturer.
3. Registration of the Medical Device:
Most medical devices require registration through the Online Medical Device Portal of CDSCO. Registration involves providing:
- Manufacturer details.
- Product details, including intended use, classification, and standards compliance.
- Technical documentation (e.g., Device Master File, Plant Master File).
4. Submission of Technical Documentation:
Manufacturers/importers must prepare and submit comprehensive documentation, which typically includes:
- Device Master File (DMF): Detailed information about the design, materials, and manufacturing processes.
- Plant Master File (PMF): Details of the manufacturing facility, including QMS compliance.
- Clinical Evaluation Report (CER): Evidence of safety and efficacy, referencing clinical trials or previous regulatory approvals.
- Risk Management Report: Based on ISO 14971, detailing risk analysis and mitigation strategies.
- Performance Benchmarks: Data demonstrating that the device meets its intended specifications.
5. Quality Management System (QMS) Compliance:
- For all medical devices, compliance with ISO 13485 (Quality Management System) is mandatory.
- The manufacturing process and facilities must adhere to these standards.
6. Clinical Investigation (If Required):
- For Class C and D devices (moderate to high risk), clinical investigations may be required if there is insufficient prior clinical data.
- Clinical trial approval is obtained from the CDSCO Clinical Trial Division.
- If clinical data from recognized global regulatory bodies (e.g., USFDA, EU CE) exists, it may suffice, reducing the need for additional trials.
7. Application for Import/Manufacturing License:
Based on the device class:
- Class A and B:
- Applications are submitted to the State Licensing Authority (SLA).
- Class C and D:
- Applications are submitted to CDSCO.
- Licensing involves submission of Form MD-3 (for manufacturing) or Form MD-14 (for imports) along with the required fees.
The CDSCO or SLA reviews the documentation and may conduct audits or inspections before granting approval.
8. Labeling and Packaging Compliance:
The device must meet Indian labeling requirements, which include:
- Name of the device.
- Manufacturer and importer details.
- Batch/lot number and expiry date.
- Instructions for use (in English).
- Unique Device Identification (UDI), if applicable.
9. Grant of License:
Upon successful evaluation, the license is granted:
- Form MD-5: Manufacturing license for Class A and B devices.
- Form MD-6: Manufacturing license for Class C and D devices.
- Form MD-15: Import license for devices manufactured abroad.
10. Post-Market Surveillance and Vigilance:
After market entry, manufacturers/importers must:
- Maintain a robust Post-Market Surveillance (PMS) system.
- Report adverse events to the Materiovigilance Programme of India (MvPI).
- Conduct periodic safety updates and risk assessments.
11. Validity and Renewal:
- Licenses are valid for five years and must be renewed before expiration.
- Regular inspections by CDSCO ensure continued compliance.
Timeline:
The approval timeline varies based on device classification and complexity:
- Class A and B devices: 6-9 months (simpler process with SLA involvement).
- Class C and D devices: 12-18 months (higher scrutiny by CDSCO).
Challenges in the Process:
- Complex documentation requirements.
- Limited number of Notified Bodies for QMS certification.
- Regulatory bottlenecks, especially for high-risk devices.
Tips for Navigating the Process:
- Engage with regulatory consultants for efficient preparation and submission.
- Leverage prior international approvals (e.g., FDA, CE mark) to expedite the process.
- Stay updated with CDSCO guidelines to ensure compliance with evolving regulations.
The approval process for bringing a medical device to market in India is rigorous but ensures the safety, efficacy, and quality of devices. Aligning with CDSCO’s regulatory framework, adhering to documentation requirements, and maintaining post-market compliance are crucial steps for a successful market entry.








{kind=link}